


URGENT: FIELD SAFETY NOTICE

Device: Relay Pro Thoracic Stent Graft System Non-Bare Stent Configuration >= diameter
32mm (28-N4 devices)
Dear Valued Customer,
The purpose of this letter is to notify consignees that Bolton Medical Inc. (subsidiary of Terumo Aortic) is conducting a voluntary medical device recall (correction) involving manufacturing changes and a labeling update for all lots of the Relay Pro Thoracic Stent Graft System, N4: Non-Bare Stent Configuration 32mm and above. Failures to release the partially deployed stent-graft from the delivery system have occurred. Please be advised that this failure mode can occur without prior warning and no device-based bailout method has been identified for this specific scenario. We advise that alternative stent-graft options be considered prior to use of the RelayPro devices impacted until the root cause evaluation is completed and effective mitigation measures are in place. In cases where the RelayPro is used and this issue is encountered, clinical judgment should guide timely decision-making, including consideration of conversion to open surgery, if appropriate.
Description of Problem:
Through post-market surveillance, we have received complaints where the graft is unable to unclasp from the delivery system because the proximal clasp is disconnected from the outer control tube. This may be recognized by the user as a lack of resistance felt when sliding the apex holder back accompanied by a failure to release the proximal stent. The implant cannot be recaptured at this stage in the procedure.
Based on our preliminary investigation, we have determined that design and manufacturing may contribute to this device problem. The root cause investigation is in process; thus, impacted devices/models and corrective actions may change.
Risk to Health:
Bolton Medical has received four (4) complaints within the last eight (8) months associated with this failure mode, three (3) of which resulted in death including one (1) aortic perforation, and two (2) conversions to open surgery which resulted in patient deaths due to stroke. In these cases, proximal clasps disconnected from the outer control tube. This prevented the stent grafts from being released from the delivery system.
This failure mode is not able to be recognized until it occurs during the procedure. Difficulties in releasing the stent graft may result in delay of procedure, stent graft displacement, and an inability to release the stent graft. This may require conversion to open surgical repair to release the clasp and can result in patient death.
Corrective Action:
In the event that there is difficulty releasing the proximal stent and no resistance is encountered when sliding the apex holder back, we recommend that the user first attempt the existing bailout techniques described in the Instructions for Use (IFU) prior to committing to any alternative course of action. If the problem persists, there are no additional device bailout methods available and clinical judgment should guide timely decision-making, including consideration of conversion to open surgery. Labeling updates will be communicated as more information becomes available.
Immediate corrective actions by Terumo Aortic have included manufacturing changes to reduce the likelihood of delivery system bond failure. Devices within the Relay Pro N4 family that incorporate support wire configurations (sizes greater or equal to 32mm diameter) have been identified as having an elevated risk of the reported failure modes due to the following:
- The investigation identified that the primary manufacturing process element leading to the failure mode is only used for the N4 NBS with support wire >= 32mm product.
- Confirmed complaints are limited to RelayPro N4 devices with support wires.
- No confirmed field failures of this type have been identified in the other product families.
At this time there is limited information to support the effectiveness of these corrective actions given the root cause investigation is still underway. Future corrective actions may include additional manufacturing and design changes.
Devices Included:
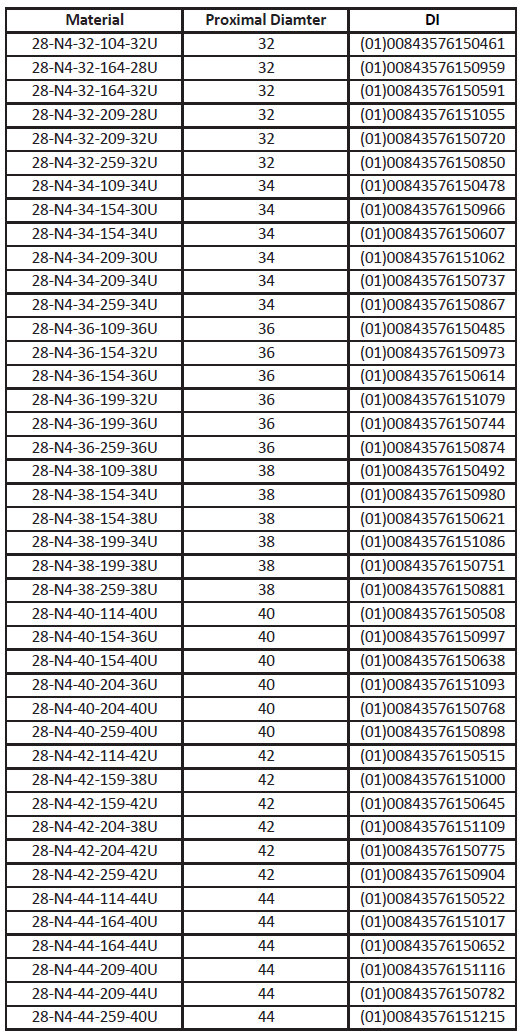
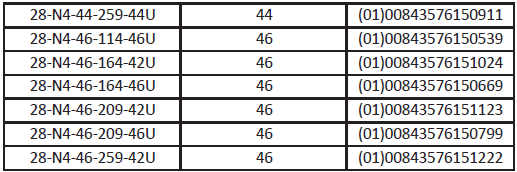
Actions To Be Taken By The Customer:
- Consider alternative stent-graft options prior to use of the RelayPro devices impacted until the root cause evaluation is completed and effective mitigation measures are in place.
- Inform all users of Relay Pro of the additional guidance for managing cases where the stent graft cannot be released from the delivery system.
- Please post a copy of this notification where the devices are stored and keep with the IFU.
- Complete Appendix 1 acknowledging receipt and communication of this medical device correction notice. Return Appendix 1 to Terumo6732@sedgwick.com.
- If devices have been transferred to another facility, please provide them with a copy of the notification and instruct them to follow the actions in this section.
Please be assured that we take the safety and quality of our products very seriously and remain committed to supporting you with clear guidance and ongoing monitoring. If you have additional questions, please reach out to your Terumo Aortic Representative or contact us Market_Actions-TMC@terumomedical.com.
Adverse Health events may be reported to the FDA's MedWatch Adverse Event Reporting program via:
Web: MedWatch website at http://www.fda.gov/medwatch
Phone: 1-800-FDA-1088 (1-800-332-1088)
Mail: MedWatch, HF-2, FDA, 5600 Fisher’s Lane, Rockville, MD 20852-9787
On behalf of Bolton Medical, Inc.
Kimberly Feitl
Vice President, Quality
URGENT: FIELD SAFETY NOTICE
Appendix 1: Medical Device Correction Notice Acknowledgement Form
FSN2026-002
Account Information:
Customer Name: |
|
Customer Address: |
|
Customer Telephone Number: |
|
Customer e-mail Address: |
|
By signing below, I acknowledge:
· Receipt of the medical device correction notice and confirm that I understand the contents.
· I have communicated the information to users in my organization.
Name (Print): |
|
Signature: |
|
Date: |
|
Please return completed form to Terumo6732@sedgwick.com.